Hemophilia A Guidelines
<
(Edição 2022)
Hemophilia A (HA) é uma desordem hemorrágica hereditária que é herdada de forma recessiva numa cadeia cromossómica X. A principal manifestação clínica é uma anomalia qualitativa ou quantitativa do factor VIII de coagulação (FVIII). A apresentação clínica é caracterizada por uma hemorragia que é difícil de parar após um trauma espontâneo ou menor nas articulações, músculos, vísceras e tecidos profundos, muitas vezes começando na infância. A incidência de HA nos homens é de aproximadamente 1 em 5.000 e a hemofilia nas mulheres é extremamente rara. A prevalência da hemofilia na China é de 2,73 por 100.000 habitantes, com HA a representar 80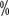 ~The prevalência da hemofilia é de 2,73 por 100.000 habitantes, com HA representando 80
~The prevalência da hemofilia é de 2,73 por 100.000 habitantes, com HA representando 80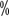 “.
“.
HA é uma doença causada por deficiência ou qualidade anormal de um único factor de coagulação. O reconhecimento precoce e diagnóstico de hemofilia permite aos doentes viver uma vida normal, evitando a hemorragia e os danos e incapacidades articulares causados pela hemorragia através de um tratamento preventivo racional e correcto, ou uma terapia de substituição atempada após a hemorragia.
Devido ao importante papel de FⅧ no caminho da coagulação endógena, pacientes com HA O quadro clínico é de hemorragia que pode ocorrer em qualquer parte do corpo. Os locais mais comuns de hemorragia são as articulações, músculos e tecidos profundos, mas também o gastrointestinal , urinary , sistema nervoso central e após a extracção dentária. hemorragia, etc. Se não for tratada, pode levar a deformidades articulares e pseudotumores, e em casos graves pode mesmo ser fatal. A doença caracteriza-se também por hemorragia persistente após trauma ou cirurgia.
O grau de sangramento está relacionado com FVIII actividade. Atividade correlaciona-se com pacientes mais leves, que geralmente sangram raramente e só após lesão ou cirurgia; pacientes mais pesados, que sangram desde uma idade precoce e podem sangrar de qualquer parte do corpo; e pacientes intermediários, que sangram mais severamente do que os tipos mais leves e mais pesados.
Uma desordem hemorrágica, incluindo a hemofilia, deve ser considerada quando um paciente do sexo masculino, especialmente uma criança, apresenta uma hemorragia espontânea, ou hemorragia que não pára após trauma ou cirurgia. Acompanhar a história familiar do paciente e completar mais testes laboratoriais para confirmar o diagnóstico.
(i) Teste de rastreio.
Incluindo contagem de plaquetas, esfregaço de sangue periférico (morfologia das plaquetas), tempo de protrombina (TP), tempo de tromboplastina parcial activada (APTT) Os pacientes com HA apresentam apenas um APTT prolongado, mas alguns pacientes com HA ligeiro têm apenas um limite ligeiramente prolongado ou elevado de APTT normal. A contagem e morfologia das plaquetas, assim como outros parâmetros de coagulação, devem ser normais.
(ii) Testes de confirmação.
Testes do factor de coagulação: Um APTT prolongado sugere um processo de coagulação endógena anormal e os marcadores de coagulação associados a isto devem ser verificados, incluindo FVIII, FIX, actividade FXII e antigénio do factor de hemofilia vascular (VWF:Ag).
VIII:C) é reduzido ou ausente, VWF:Ag é normal, e FVIII:C/VWF:Ag é acentuadamente reduzido, sugerindo HA.
(iii) Ensaio inibidor.
1.
Patientes com HA devem ser testados para inibidores FⅧ quando são menos eficazes do que antes, e antes de serem submetidos a cirurgia. Em pacientes pediátricos, recomenda-se a realização de testes a cada 5 dias de exposição durante os primeiros 20 dias após o primeiro tratamento com um produto F VIII e a cada 21-50 dias de exposição após o primeiro tratamento com um produto F VIII.
cada 10 dias de exposição durante os primeiros 20 dias de exposição e pelo menos 2 testes por ano a seguir.
até 150 dias de exposição. 2. rastreio inibidor.
O teste de correcção APTT é realizado utilizando uma mistura 1:1 de plasma normal e plasma do paciente, e o APTT é medido imediatamente e novamente após 2 horas de incubação a 37°C, e comparado com o APTT de sujeitos normais e do próprio paciente. Se a incubação durante 2 horas não corrigir, é indicada a presença de um inibidor do factor de coagulação (ver Quadro 1 para a interpretação do teste de correcção APTT).
3.
Os títulos dos inibidores devem ser medidos para confirmar um diagnóstico de inibidor. Se se descobrir que o paciente tem um título inibidor ≥ 0,6 BU/ml em 2 ocasiões consecutivas dentro de 1 a 4 semanas pelo método Bethesda ou Nijmegen, o paciente é considerado positivo. Se o título inibidor é >5 BU/ml, o título inibidor é alto; se o título inibidor é ≤5 BU/ml, o título inibidor é baixo.
(iv) Genetic testing.
Testes genéticos de pacientes são recomendados para identificar o gene causal e fornecer uma base para testes de portadores e diagnóstico pré-natal na mesma família. Além disso, as mutações podem ser utilizadas para determinar o risco do paciente de desenvolver o supressor.
Segundo o nível de actividade de FⅧ, HA pode ser classificado em tipos leves, moderados e pesados (ver Os três tipos de HA são leves, moderados e pesados (ver Quadro 2). Atividade fatorial <1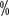 é considerada pesada; atividade 1
é considerada pesada; atividade 1 a 5
a 5  para meio; actividade >5
para meio; actividade >5  a 40
a 40 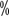 é leve.
é leve.
V. Diagnóstico e diagnóstico diferencial
(i) Diagnóstico.
O diagnóstico foi baseado na apresentação clínica do paciente com hemorragias graves recorrentes desde a infância, especialmente nas articulações, combinado com FVIII factor O diagnóstico pode ser confirmado por testes laboratoriais para diminuição da actividade e antigénio com mutações no gene F VIII.
(ii) Diagnóstico diferencial.
<
- Hemofilia vascular (doença de von Willebrand): A VWD é uma desordem autossómica causada por um defeito qualitativo ou quantitativo na vWF. É maioritariamente herdada de forma dominante. Os pacientes têm tendência a sangrar, mas predominantemente a partir da pele e das mucosas. Porque o vWF tem a capacidade de aumentar a estabilidade de FVIII:C, prevenir a sua degradação e promover a sua produção e libertação, os pacientes com VWD podem desenvolver FVIII actividade diminuída. O diagnóstico e o estadiamento do VWD requer testes de antigénios e actividade vWF (actividade de cofactor de ristocetina, actividade vWF). O VWD pode ser diagnosticado e tipado com antigénio vWF e actividade (actividade do cofactor ristomicina, vWF R:Co), ensaio de ligação de colagénio, FVIII ensaio de ligação, ensaio de adesão e agregação de plaquetas, electroforese de proteínas vWF, etc. Electroforese de proteínas vWF, etc. O diagnóstico genético é também um instrumento de diagnóstico.
-
HA adquirido: é a presença de anti-F<VIII style=”font-family:Arial”>autoanticorpos causadores de FVIIIuma forma de doença auto-imune que requer diferenciação de HA, especialmente em pacientes com HA combinados com supressores. O HA adquirido caracteriza-se por não ter antecedentes de hemorragia ou historial familiar positivo e ocorre mais frequentemente em doentes com malignidade, doença auto-imune e mulheres perinatais, embora cerca de metade dos doentes não tenha um desencadeamento aparente e a terapia imunossupressora seja eficaz.
/p>
- >
-
Outras deficiências de factores de coagulação herdados: Os doentes que se descobriu terem apenas um APTT prolongado devem também ser testados para a actividade dos factores de coagulação como F Ⅸ, F Ⅺ, F Ⅻ, F Ⅴ e F Ⅹ que podem causar APTT prolongados ao realizar testes confirmatórios para excluir as correspondentes deficiências de factores de coagulação.
<(i) Princípios de tratamento.
Patientes com HA requerem terapia de substituição com FⅧ e terapia de substituição regular (profilaxia) na ausência de hemorragia, com o objectivo de parar a hemorragia e assim maximizar a protecção da função articular. O objectivo é parar a hemorragia de modo a maximizar a função articular, dar um tratamento adequado a pedido em caso de hemorragia, e dar uma terapia de substituição adequada em caso de cirurgia ou outras operações traumáticas. Os pacientes com HA devem evitar injecções intramusculares e traumas.
(ii) Terapia de substituição.
- style=”margin-left: 48pt”>
- Selecção de fármacos de terapia alternativa.
Recombinante F VIII ou F VIII derivado do sangue viralmente inactivado é preferível para a terapia de substituição de HA, mas a precipitação a frio ou plasma fresco congelado pode ser usado se estes fármacos não estiverem disponíveis. Cada 1 UI/kg de peso corporal de FVIII infusão pode levar a um aumento de FVIII actividade no corpo.
(FⅧ:C) por 2% e o FVIII no corpo é de 8-12 horas, e é importante manter o FVIII:C no corpo a um certo nível. :C deve ser infundido a cada 8 a 12 horas para manter um certo nível.
FVIII primeiro requisito = (FVIII infusão necessária para alcançar Roman”>Ⅷ concentração– basal do paciente FⅧ concentração F× peso (kg)× 0,5; após a primeira dose, metade da primeira dose pode ser infundida a cada 8 a 12 horas, conforme apropriado A primeira dose pode ser administrada a cada 8-12 horas até se conseguir uma hemostasia completa.
As taxas de recuperação e meia-vida variam consideravelmente entre indivíduos, e recomenda-se que as unidades com capacidade para o fazer revejam a dose.
- Selecção de fármacos de terapia alternativa.
Medir parâmetros farmacocinéticos tais como recuperação e meia-vida em pacientes e orientar a terapia com base nos resultados.
- style=”margin-left: 59pt”>
- Implantação de tratamentos alternativos.
>p>O tratamento alternativo é dividido em tratamento a pedido e tratamento alternativo regular (tratamento preventivo).
-
Tratamento a pedido: refere-se ao tratamento de pacientes com hemorragia aguda ou quando a corrente mais eficaz A medida hemostática mais eficaz continua a ser FVIIIterapia de substituição, com os princípios de tratamento precoce, dose adequada e duração do tratamento. Terapia de substituição FVIII style=”font-family:Arial”>dose e duração devem ter em conta o local e a gravidade da hemorragia (ver Tabela 3).
-
Terapia de substituição perioperatória: refere-se à terapia de substituição administrada antes, durante e após a cirurgia. O objectivo é assegurar o êxito da cirurgia e a recuperação dos pacientes com HA. Ver Quadro 4 para detalhes de opções de tratamento alternativas.
< -
Tratamento profiláctico: Esta é a terapia de substituição regular dada regularmente para prevenir hemorragias. Como o tratamento on-demand é apenas pós-casamento e não pode prevenir hemorragias recorrentes que conduzem a incapacidade articular em doentes com AH grave, o tratamento profiláctico é particularmente crítico uma vez que o objectivo é manter a função articular e muscular normal. O tratamento profiláctico é o tratamento de escolha para crianças com HA. Deve ser estabelecido um objectivo de menos de três hemorragias articulares por ano para os doentes pediátricos, a fim de minimizar a ocorrência de danos articulares e incapacidade articular irreversível devido a hemorragias articulares.
Não há consenso sobre a adesão à profilaxia em pacientes adultos, mas a experiência nacional e internacional demonstrou que a profilaxia terciária de curto prazo pode reduzir o número de hemorragias e melhorar a qualidade de vida. Além disso, para pacientes que sofreram recentemente um aumento da hemorragia, particularmente na articulação alvo, recomenda-se um curto curso de profilaxia de 4-8 semanas para interromper o ciclo vicioso de danos na articulação de sangramento. Este tratamento pode ser combinado com fisioterapia intensiva ou radioterapia.
Sinovectomia de radiação.
Há normalmente três tipos de tratamento profiláctico: 1) profilaxia primária: após o diagnóstico, antes da segunda hemorragia articular e quando a criança tem menos de 3 anos de idade e não há provas claras
Terapia de substituição regular em curso é iniciada quando a presença de artropatia é confirmada (no exame ou imagem); (ii) Terapia de prevenção secundária: A terapia de substituição regular em curso é iniciada após dois ou mais episódios de hemorragia na articulação, mas não é encontrada artropatia no exame e/ou imagem; (iii) Terapia de prevenção terciária: A terapia de substituição regular em curso é iniciada após dois ou mais episódios de hemorragia na articulação. (3) Profilaxia terciária: a terapia de substituição contínua regular só é iniciada após a presença de uma artropatia ter sido confirmada no exame e imagiologia.
Para prevenir o aparecimento de incapacidade articular, recomenda-se que a profilaxia seja iniciada em crianças com doença grave logo que ocorra a primeira hemorragia articular, hemorragia muscular grave, hemorragia intracraniana ou outra hemorragia que ponha em risco a vida. As crianças com historial de hemorragias articulares e artropatia devem começar a profilaxia cedo, dependendo do seu estado, e visar um número anual de hemorragias articulares ou <3 hemorragias por ano, se possível.

③Small regime de dose: 10-15 UI/kg por dose, 2 a 3 vezes por semana; seguido de
30 IU/kg duas vezes por semana; seguido de 25 IU/kg por dose de dois em dois dias. A dose e a frequência do tratamento podem ser optimizadas e os recursos podem ser atribuídos de forma a garantir a eficácia em comparação com os regimes de altas doses.
Embora o regime profiláctico óptimo ainda esteja por determinar, um regime de baixas doses pode reduzir significativamente a hemorragia em crianças com hemofilia em comparação com o tratamento a pedido, mas não reduz a incidência de artropatia. Recomenda-se que seja implementado um regime de profilaxia de dose média em crianças com hemofilia financeiramente capazes de o fazer, ou que seja desenvolvido um regime individualizado óptimo com base na idade, acesso venoso, fenótipo hemorrágico, características farmacocinéticas e disponibilidade de preparações de factores de coagulação.
(iii) Non-factor therapy.
<
-
Emicizumab: um anticorpo monoclonal bisespecífico que imita F. família:Times New Roman”>VIIIa imitando a função de cofactor de FVIII>a e fazendo a ponte entre a função de cofactor de FVIIIa. span style=”font-family:Arial”>a e FX, permitindo F style=”font-family:Arial span style=”font-family:Times New Roman”>Xna ausência de FVIII style=”font-family:Times New Roman”>. span>< pode continuar a ser activado na ausência de F, restaurando a via de coagulação fisiológica. É aprovado para o tratamento profiláctico de rotina de pacientes com HA combinado com FVIIIinibidores na China e também nos EUA e UE sem F style=”font-family:Arial”>inibidores. span><VIIIinibidores, e nos EUA e UE para tratamento profiláctico de rotina de pacientes HA sem Finibidores. O regime de dosagem recomendado é uma dose de carga de 3 mg/kg administrada subcutaneamente uma vez por semana durante as primeiras 4 semanas para atingir rapidamente as concentrações de sangue alvo e uma dose de 3 mg/kg durante as primeiras 4 semanas.
Uma dose de manutenção de 1,5 mg/kg é dada uma vez por semana a partir da semana 5.
<
-
Deamino-8-D-arginine booster (DDAVP): HA leve O DDAVP é opcional em pacientes com hemorragia e pode ser eficaz em alguns HAs intermédios, mas não em pacientes com HA pesados. A dose recomendada é de 0,3 a 0,4μg/kg, diluído em 50 ml de soro fisiológico e administrado lentamente por via intravenosa (pelo menos 30 minutos) a cada 12 horas. A cada 12 horas durante 1 a 3 dias. Aumento da concentração do factor de coagulação >30
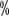 ou mais após administração
ou mais após administração> >3 vezes mais eficaz. O medicamento é ineficaz após múltiplas doses e deve ser suplementado com FVIII preparações em caso de maus resultados. As reacções adversas incluem a lavagem temporária e a retenção de sódio. Devido a reacções adversas como a retenção de sódio e água, o
Contraindicado em crianças com menos de 2 anos de idade. A restrição da água e os pré-testes são necessários para utilização em crianças pequenas. As crianças pré-testadas também podem ser tratadas com spray nasal DDAVP especificamente para hemofilia, para controlar pequenas hemorragias.
>ul><
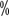 de solução de ácido tranexâmico 10ml Lavar durante 2 minutos, 4 vezes por dia durante 7 dias.
de solução de ácido tranexâmico 10ml Lavar durante 2 minutos, 4 vezes por dia durante 7 dias.(iv) Fisioterapia.
Os pacientes são encorajados a fazer exercícios aeróbicos apropriados e seguros (natação, ciclismo motorizado, jogging, caminhada rápida, etc.) com treino apropriado de resistência à carga e auto-alongamento durante períodos sem hemorragia para prevenir e reduzir a recorrência de hemorragias.
A gestão da hemorragia deve seguir os princípios de PREÇO de Proibição, Descanso, Gelo, Compressão, Elevação, etc.
(Elevação). Em caso de hemorragia muscular e articular, o princípio PRICE é dar ao paciente uma compressa fria antes de infundir a coagulação.
O princípio PREÇO é uma importante medida de gestão baseada na infusão de factores de coagulação para elevar os níveis de factores de coagulação, e o uso atempado de talas, moldes, muletas ou A travagem com talas, moldes, muletas ou cadeiras de rodas pode colocar os músculos e articulações em posição de repouso e a utilização de gelo ou pacotes molhados a frio pode ser eficaz na redução da resposta inflamatória. Recomenda-se a aplicação de gelo a cada 4-6 horas durante cerca de 5-10 minutos (não mais do que 10 minutos de cada vez) até que o inchaço e a dor sejam reduzidos.
Além disso, um profissional de reabilitação/terapeuta treinado pode avaliar o paciente em termos de função dos membros, mobilidade individual e participação social e, com base nos resultados da avaliação, orientar o paciente através de exercícios de reabilitação para prevenir, reduzir e minimizar disfunções musculares e articulares e melhorar a vida diária. Os resultados da avaliação são utilizados para orientar o paciente através de exercícios de reabilitação para prevenir, reduzir e minimizar disfunções musculares e melhorar a actividade diária e a qualidade de vida.
<(a) Tratamento de inibidores complicadores.
Patientes com HA recebendo FVIII terapia de substituição produz anticorpos homo-neutralizantes chamados supressores, e a taxa de produção de supressores em doentes com HA pesado é de 20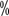 a 30
a 30 , e cerca de 5 para média ou leve HA
, e cerca de 5 para média ou leve HA a 10
a 10  Possibilidade de produzir inibidores. A presença contínua de inibidores é uma complicação grave da hemofilia, levando a sintomas de hemorragia mais difíceis de controlar, aumento do risco de hemorragia fatal e uma maior redução na qualidade de vida. A presença de inibidores é tratada tanto como tratamento hemostático como como remoção de inibidores.
Possibilidade de produzir inibidores. A presença contínua de inibidores é uma complicação grave da hemofilia, levando a sintomas de hemorragia mais difíceis de controlar, aumento do risco de hemorragia fatal e uma maior redução na qualidade de vida. A presença de inibidores é tratada tanto como tratamento hemostático como como remoção de inibidores.
- style=”margin-left: 59pt”>
- Tratamento demostático.
>p>Patientes que estão a sangrar gravemente devem ser tratados o mais depressa possível para estancar a hemorragia. Existem várias formas de hemostasia.
-
High dose FVIII: utilização apenas em pacientes com hemorragia HA combinada com inibidores de baixo título (≤5 BU/ml). A quantidade de FVIII requerida inclui a quantidade utilizada para neutralizar o inibidor e a quantidade necessária para parar a hemorragia. A quantidade de FVIII style=”font-family:Arial”> utilizada para neutralizar o inibidor é calculada
O método é o seguinte: Peso (kg)
× 80 × [(1 – volume específico do eritrócito) x título inibidor (BU)]. A isto são necessárias 50 UI/Kg adicionais de FⅧ para assegurar que FⅧ:C possa ser detectado in vivo. Aumentar. Se a hemostasia não for eficaz, aumentar a dose ou encurtar o intervalo de dosagem, ou mudar para a terapia de bypass. No caso de inibidores de baixo teor, altamente reactivos (título inibidor >5 BU/ml após uma segunda infusão de FVIII), considerar mudar para a via de derivação após 3 a 5 dias de dosagem.
<
- Preparações de desvio: para inibidores combinados de alto título (>5 BU/ml) ou indução de tolerância imunológica A via do bypass é indicada para pacientes que falharam o tratamento com inibidores de títulos elevados (>5 BU/ml) ou indução de tolerância imunológica (ITI), ou que estão a sangrar com ITI. Drogas alternativas de “via de desvio” incluem concentrado de complexo de protrombina activado (aPCC) e factor VII de coagulação activo recombinante (rF VIIa). rFⅦa O método de administração é 90μg/kg intravenoso a cada 2-4 horas ou 270. style=”font-family:Times New Roman”>μg/kg dado como dose única. A dose recomendada de PCC é de 50-100 U/kg por dose, a intervalos de 8-12 horas, para não exceder 200 U/kg por dia. Uma vez estabilizada a hemorragia aguda, o tratamento profilático com PCC ou rFⅦa é necessário durante pelo menos 6 meses.
-
Emicizumab: profilaxia de Emicizumab Tem demonstrado ser útil no controlo da hemorragia, no restabelecimento da função das articulações-alvo e na melhoria da qualidade de vida dos hemofílicos. O regime de dosagem é o mesmo descrito acima, com as preparações de bypass descontinuadas 24 horas antes da dosagem. Se ocorrer uma hemorragia revolucionária durante a profilaxia, rFVIIa deve ser usado como primeira escolha de tratamento, com uma dose inicial de ≤90. μ style>g/kg quando administrado repetidamente em intervalos de tratamento
deve ser superior a 2 horas. Também para evitar tromboses, os medicamentos aPCC ou PCC devem ser evitados. FⅧ também pode ser usado para tratar hemorragias de ruptura em pacientes com inibidores combinados de baixo teor de inibidores.
- style=”margin-left: 59pt”>
- Remoção de inibidores.
ITI é o único método actualmente aceite de eliminação de inibidores em pacientes inibidores-positivos que foram tratados regularmente e frequentemente com preparações de factores de coagulação durante um longo período de tempo para alcançar a tolerância imunitária periférica.
-
Timulação da iniciação de ITI: Não existe consenso internacional sobre quando é a melhor altura para iniciar a ITI, mas a preferência actual é começar uma vez feito o diagnóstico, independentemente do título inibidor. O ITI deve ser iniciado assim que o diagnóstico for confirmado, independentemente do título do inibidor.
< -
Opção da preparação do factor de coagulação: F derivada do sangue>. span><VIII style=”font-family:Arial”> preparações concentradas ou rhFVIIIestão disponíveis, não há evidências que sugiram qual a formulação superior, mas quando se usa rhFVIII style=”font-family:Arial”> para o tratamento ITI não é bem sucedido, pode ser considerada uma mudança para FVIIIconcentrate :Arial”>Preparações concentradas.
<
>ITI regime: i) Tratamento de primeira linha:iDose elevada: 200 UI/(kg style=”font-family:Times New Roman”>- d); style=”font-family:equivocal; font-size:10pt”>ii Medium dose: 100 IU/(kg–>d); iii Dose baixa: 25-50 UI/kg uma vez em cada dois dias ou 3 vezes por semana. Não houve diferença no sucesso da indução de tolerância imunológica no grupo de alta dose em comparação com o grupo de baixa dose, mas o grupo de alta dose teve um início de acção mais rápido e uma redução significativa dos sintomas de hemorragia durante o tratamento. Além disso, a dose baixa de ITI pode ser combinada com o emicizumabe. Verificou-se que isto reduz a quantidade de factores de coagulação e evita hemorragias frequentes. Uma vez iniciado um ITI, este não deve ser descontinuado à vontade, uma vez que isto pode afectar a eficácia dos ITI subsequentes. Após iniciar uma ITI, os títulos inibidores devem ser testados uma vez por semana e se
Os títulos inibidores não aumentados ou uma diminuição dos títulos inibidores de menos de 20% durante seis meses devem levar a um aumento gradual da dose de ITI até 200 IU/(kg–d); se a dose tiver atingido 200 IU/(kg–d), recomenda-se uma mudança para um regime de segunda linha. (ii) Terapia de segunda linha: Não existe uma terapia de segunda linha padrão, considere mudar para um estilo FⅧ produto, por exemplo de rhFⅧ para FVIII e também em combinação com inibidores de eliminação de anticorpos monoclonais CD20 derivados de humanos, mas é necessária uma avaliação mais aprofundada da eficácia e segurança a longo prazo.
- style=”margin-left: 83pt”>
-
Critérios para avaliar a eficácia do ITI: ① Totalmente tolerado. Inibidor negativo persistente
(<0,6 BU/ml) e FⅧ recuperação >66%, FVIII meia-vida > 6 horas; (ii) parcialmente tolerado: títulos inibidores <5 BU/ml, embora FVIII recuperação seja inferior a 66% e/ou meia-vida < 6 horas, mas o tratamento com FⅧ pode parar a hemorragia; (iii) Ineficaz: a tolerância total ou parcial não pode ser alcançada. Geralmente, os títulos de inibidores caem menos de 20% dentro de 3-6 meses ou permanecem >5 BU/ml após 3-5 anos de ITI.
-
Previsão da eficácia de ITI: Considera-se actualmente que a eficácia de ITI é provavelmente melhor em pacientes com as seguintes características: 1) título inibidor <10 BU/ml antes do início de ITI (ii) títulos inibidores de pico históricos <10 BU/ml antes do início da ITI; (iii) títulos inibidores de pico <100 BU/ml durante a ITI; (iv) tempo desde o diagnóstico até ao início da ITI <5 anos; e (v) sem interrupções desde o início da ITI.
E os resultados da ITI podem ser mais pobres em pacientes com: i) títulos inibidores ≥10 BU/ml antes de iniciar a ITI; ii) títulos inibidores de pico históricos ≥200 BU/ml; iii) títulos inibidores de pico >100 BU/ml durante a ITI; iv) tempo desde o diagnóstico até ao início da ITI >5 anos. (iv) >5 anos desde o diagnóstico até ao início da ITI; (v) >2 semanas de intervalo após o início da ITI.
- style=”margin-left: 72pt”>
-
Timulação do fim do ITI: aqueles que atingiram a tolerância total Mudar para profilaxia
-
Tratamento para aqueles que atingiram tolerância total; para aqueles que atingiram tolerância parcial, se F
 ou se a terapia ITI não tiver sido total ou parcialmente tolerada durante 3 a 5 anos.
ou se a terapia ITI não tiver sido total ou parcialmente tolerada durante 3 a 5 anos.


 >
>